Enguerrand.github.io
CC1 ADM
Enguerrand Appy-Ruat
Ce script s’appuie sur le “DADA2 Pipeline Tutorial (1.16) et l’analyse des données MiSeq_SOP y étant associées.
Chargement du package dada2
library(dada2)
Vérification de la version du package
packageVersion("dada2")
Ouverture du fichier de données
path <- "~/MiSeq_SOP"
list.files(path)
##[1] "F3D0_S188_L001_R1_001.fastq" "F3D0_S188_L001_R2_001.fastq"
##[3] "F3D1_S189_L001_R1_001.fastq" "F3D1_S189_L001_R2_001.fastq"
##[5] "F3D141_S207_L001_R1_001.fastq" "F3D141_S207_L001_R2_001.fastq"
##[7] "F3D142_S208_L001_R1_001.fastq" "F3D142_S208_L001_R2_001.fastq"
##[9] "F3D143_S209_L001_R1_001.fastq" "F3D143_S209_L001_R2_001.fastq"
##[11] "F3D144_S210_L001_R1_001.fastq" "F3D144_S210_L001_R2_001.fastq"
##[13] "F3D145_S211_L001_R1_001.fastq" "F3D145_S211_L001_R2_001.fastq"
##[15] "F3D146_S212_L001_R1_001.fastq" "F3D146_S212_L001_R2_001.fastq"
##[17] "F3D147_S213_L001_R1_001.fastq" "F3D147_S213_L001_R2_001.fastq"
##[19] "F3D148_S214_L001_R1_001.fastq" "F3D148_S214_L001_R2_001.fastq"
##[21] "F3D149_S215_L001_R1_001.fastq" "F3D149_S215_L001_R2_001.fastq"
##[23] "F3D150_S216_L001_R1_001.fastq" "F3D150_S216_L001_R2_001.fastq"
##[25] "F3D2_S190_L001_R1_001.fastq" "F3D2_S190_L001_R2_001.fastq"
##[27] "F3D3_S191_L001_R1_001.fastq" "F3D3_S191_L001_R2_001.fastq"
##[29] "F3D5_S193_L001_R1_001.fastq" "F3D5_S193_L001_R2_001.fastq"
##[31] "F3D6_S194_L001_R1_001.fastq" "F3D6_S194_L001_R2_001.fastq"
##[33] "F3D7_S195_L001_R1_001.fastq" "F3D7_S195_L001_R2_001.fastq"
##[35] "F3D8_S196_L001_R1_001.fastq" "F3D8_S196_L001_R2_001.fastq"
##[37] "F3D9_S197_L001_R1_001.fastq" "F3D9_S197_L001_R2_001.fastq"
##[39] "filtered" "HMP_MOCK.v35.fasta"
##[41] "Mock_S280_L001_R1_001.fastq" "Mock_S280_L001_R2_001.fastq"
##[43] "mouse.dpw.metadata" "mouse.time.design"
##[45] "stability.batch" "stability.files"
[41] "Mock_S280_L001_R1_001.fastq" "Mock_S280_L001_R2_001.fastq"
[43] "mouse.dpw.metadata" "mouse.time.design"
[45] "stability.batch" "stability.files"
La fonction path sert à localiser le fichier, puis list.files va permettre d’ouvrir le fichier et vérifier la présence de nos données.
Création des objets fnFs et fnRs, listes des fichiers Forward et Reverse
fnFs <- sort(list.files(path, pattern="_R1_001.fastq", full.names = TRUE))
fnRs <- sort(list.files(path, pattern="_R2_001.fastq", full.names = TRUE))
sample.names <- sapply(strsplit(basename(fnFs), "_"), `[`, 1)
La fonction Sort permet un tri par ordre alphabétique, puis path, pattern=”_R1_001.fastq” va retenir tous les fichiers contenant “_R1_001.fastq” dans le dossier “path”
Récupération des fichiers Forward(R1) et Reverse(R2), puis extraction des noms des échantillons.
Visualisation de qualité des lectures
Forward
plotQualityProfile(fnFs[1:2])
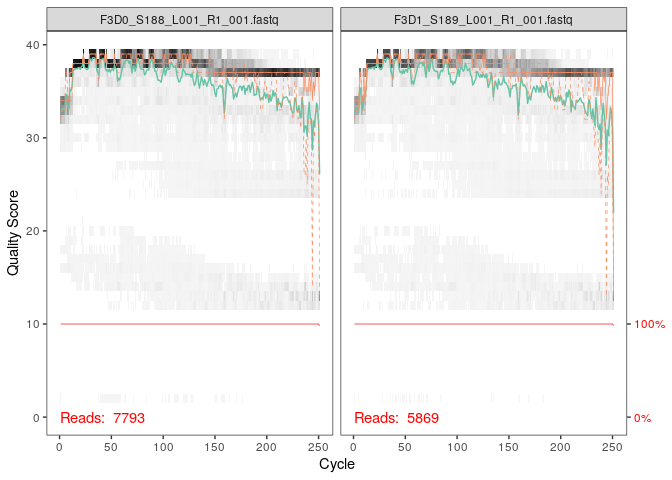
Ceci est un graphique permettant de constater la qualité des nucléotides à travers les lectures.
Le dégradé de noir représente la fréquence de score pour les différentes positions de lecture
La ligne verte es la moyenne du score de qualité pour chacune des positions.
La ligne orange (du haut) correspond au quartiles.
La ligne rouge représente la proportion de lecture qui atteignent bel et bien les différenets positions.
La lecture est ici de bonne qualité jusqu’à environs 240 paire de bases.
Reverse
plotQualityProfile(fnRs[1:2])
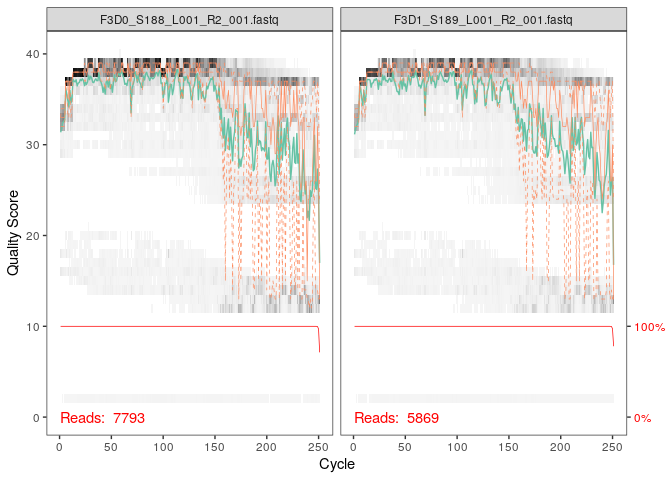
La qualité des lectures Reverse est bien plus basse que celle des lectures forward. En effet, une chute de cette qualité peut être observée à partir d’environ 155 paire de bases. Cela n’est pas étonnant pour des données Illumina, mais dada2 présente un avantage vis à vis de cela en intégrant des informations de bonne qualité dans son modèle d’erreur, compensant donc ces séquences de “mauvaise” qualité.
Nommer fichiers filtrés
filtFs <- file.path(path, "filtered", # Crée un chemin vers un sous-dosier "filtered" à l'intérieur du dossier 'path"
paste0(sample.names, "_F_filt.fastq.gz")) # Crée le nom du fichier filtré pour chaque échantillon
filtRs <- file.path(path, "filtered", paste0(sample.names, "_R_filt.fastq.gz"))
names(filtFs) <- sample.names
names(filtRs) <- sample.names # Attribution de ces noms aux fichiers
Cette étape permet en premier lieu de créer un nouveau dossier “filtered” et de créer des noms à ces fichiers filtrés ; avant de leur attribuer.
Nos fichiers sont donc maintenant reconnaissables et leur nom est lié avec celui de leur échantillon.
Filtrage des séquences
out <- filterAndTrim(fnFs, filtFs, fnRs, filtRs, truncLen=c(240,160), # Tronque les lectures Forward à 240 bases et Reverse à 160 bases
maxN=0, # Elimine toutes lecture contenant une base inconnue
maxEE=c(2,2), # Maximum d'erreurs par lecture
truncQ=2, # Tronque la lecture dès que son score de qualité est inférieur à 2
rm.phix=TRUE, # Supprime les lectures provenant du génome PhiX
compress=TRUE, multithread=FALSE)
head(out)
reads.in reads.out
F3D0_S188_L001_R1_001.fastq 7793 7113
F3D1_S189_L001_R1_001.fastq 5869 5299
F3D141_S207_L001_R1_001.fastq 5958 5463
F3D142_S208_L001_R1_001.fastq 3183 2914
F3D143_S209_L001_R1_001.fastq 3178 2941
F3D144_S210_L001_R1_001.fastq 4827 4312
Cette ligne de code permet de nous montrer les lectures avant (reads.in) et après filtrage (reads.out). Cela nous permet de constater que la majorité des lectures ont été conservées malgrès le filtrage.
Modèle d’erreur
Dada2 se base sur un modèle d’erreur paramétrique, qui lui permettra de distinguer les vrais bases (séquences biologiques) des erronées.
A préciser que la fonction “multithread=False”, écrite à de nombreuses reprise à partir de cette partie, permet d’utiliser plusieurs coeurs à la fois afin d’aller plus rapidement. Elle n’est pas utilisée ici car déconseilée sur Windows.
errF <- learnErrors(filtFs, multithread=FALSE)# Modèle statistique des erreurs à partir des lectures filtrées Forward
## 33514080 total bases in 139642 reads from 20 samples will be used for learning the error rates.
errR <- learnErrors(filtRs, multithread=FALSE)# Modèle statistique des erreurs à partir des lectures filtrées Reverse
## 22342720 total bases in 139642 reads from 20 samples will be used for learning the error rates.
Affichage du taux d’erreur
plotErrors(errF, nominalQ=TRUE)
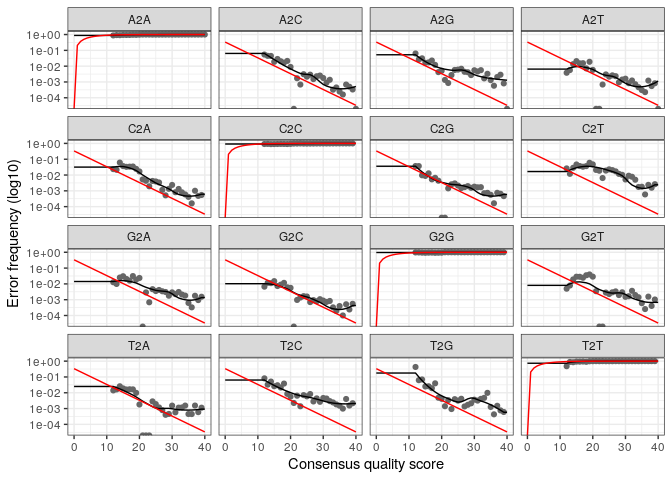
Ce graphique représente le score de qualité par rapport à la fréquence d’erreur.
Les points et lignes noires correspondent respectivement aux taux d’erreurs observés et au taux d’erreur estimé ; tandis que la ligne rouge représente le taux d’erreur théorique (selon le Q score).
Les points et lignes noires corroborent dans la plus grande majorité des cas, signifiant que le taux d’erreur observé et celui estimé sont pratiquement les mêmes.
Application de l’algorithme du modèle d’erreur de dada2
dadaFs <- dada(filtFs, err=errF, multithread=FALSE)
L’algorhithme de dada2 va comparer les fichiers forward filtrés précédemment grâce aux modèles d’erreurs incuclqués dans l’étape précédente.
## Sample 1 - 7113 reads in 1979 unique sequences.
## Sample 2 - 5299 reads in 1639 unique sequences.
## Sample 3 - 5463 reads in 1477 unique sequences.
## Sample 4 - 2914 reads in 904 unique sequences.
## Sample 5 - 2941 reads in 939 unique sequences.
## Sample 6 - 4312 reads in 1267 unique sequences.
## Sample 7 - 6741 reads in 1756 unique sequences.
## Sample 8 - 4560 reads in 1438 unique sequences.
## Sample 9 - 15637 reads in 3590 unique sequences.
## Sample 10 - 11413 reads in 2762 unique sequences.
## Sample 11 - 12017 reads in 3021 unique sequences.
## Sample 12 - 5032 reads in 1566 unique sequences.
## Sample 13 - 18075 reads in 3707 unique sequences.
## Sample 14 - 6250 reads in 1479 unique sequences.
## Sample 15 - 4052 reads in 1195 unique sequences.
## Sample 16 - 7369 reads in 1832 unique sequences.
## Sample 17 - 4765 reads in 1183 unique sequences.
## Sample 18 - 4871 reads in 1382 unique sequences.
## Sample 19 - 6504 reads in 1709 unique sequences.
## Sample 20 - 4314 reads in 897 unique sequences.
L’algorithme va donc corriger les séquences présentant des différences faibles, pouvant être considérées comme des erreurs de séquençage. Si une différence est trop importante, il va la considérer comme une “amplicon sequence variant” ou ASV, =donc une séquence biologique.
dadaRs <- dada(filtRs, err=errR, multithread=FALSE)
## Sample 1 - 7113 reads in 1660 unique sequences.
## Sample 2 - 5299 reads in 1349 unique sequences.
## Sample 3 - 5463 reads in 1335 unique sequences.
## Sample 4 - 2914 reads in 853 unique sequences.
## Sample 5 - 2941 reads in 880 unique sequences.
## Sample 6 - 4312 reads in 1286 unique sequences.
## Sample 7 - 6741 reads in 1803 unique sequences.
## Sample 8 - 4560 reads in 1265 unique sequences.
## Sample 9 - 15637 reads in 3414 unique sequences.
## Sample 10 - 11413 reads in 2522 unique sequences.
## Sample 11 - 12017 reads in 2771 unique sequences.
## Sample 12 - 5032 reads in 1415 unique sequences.
## Sample 13 - 18075 reads in 3290 unique sequences.
## Sample 14 - 6250 reads in 1390 unique sequences.
## Sample 15 - 4052 reads in 1134 unique sequences.
## Sample 16 - 7369 reads in 1635 unique sequences.
## Sample 17 - 4765 reads in 1084 unique sequences.
## Sample 18 - 4871 reads in 1161 unique sequences.
## Sample 19 - 6504 reads in 1502 unique sequences.
## Sample 20 - 4314 reads in 732 unique sequences.
Ici, comme précédemment, l’algorithme va corriger les séquences des fichiers filtrés Reverse
dadaFs[[1]]
## dada-class: object describing DADA2 denoising results
## 128 sequence variants were inferred from 1979 input unique sequences.
## Key parameters: OMEGA_A = 1e-40, OMEGA_C = 1e-40, BAND_SIZE = 16
Par exemple, pour le premier échantillon des séquences Forward, l’algorithme a pu corriger des séquences pour définir 128 séquences biologiques parmi les 1979 séquences de base.
Fusion des lectures
L’objectif de cette étape est de fusionner ensemble les fichiers filtrés précédemment avec les séquences biologiques qui y ont été identifiées.
mergers <- mergePairs(dadaFs, filtFs, dadaRs, filtRs, verbose=TRUE)
Ici, la fonction “verbos=TRUE” permet d’afficher les détails pendant l’exécution des lignes de codes.
## 6540 paired-reads (in 107 unique pairings) successfully merged out of 6891 (in 197 pairings) input.
## 5028 paired-reads (in 101 unique pairings) successfully merged out of 5190 (in 157 pairings) input.
## 4986 paired-reads (in 81 unique pairings) successfully merged out of 5267 (in 166 pairings) input.
## 2595 paired-reads (in 52 unique pairings) successfully merged out of 2754 (in 108 pairings) input.
## 2553 paired-reads (in 60 unique pairings) successfully merged out of 2785 (in 119 pairings) input.
## 3646 paired-reads (in 55 unique pairings) successfully merged out of 4109 (in 157 pairings) input.
## 6079 paired-reads (in 81 unique pairings) successfully merged out of 6514 (in 198 pairings) input.
## 3968 paired-reads (in 91 unique pairings) successfully merged out of 4388 (in 187 pairings) input.
## 14233 paired-reads (in 143 unique pairings) successfully merged out of 15355 (in 352 pairings) input.
## 10528 paired-reads (in 120 unique pairings) successfully merged out of 11165 (in 278 pairings) input.
## 11154 paired-reads (in 137 unique pairings) successfully merged out of 11797 (in 298 pairings) input.
## 4349 paired-reads (in 85 unique pairings) successfully merged out of 4802 (in 179 pairings) input.
## 17431 paired-reads (in 153 unique pairings) successfully merged out of 17812 (in 272 pairings) input.
## 5850 paired-reads (in 81 unique pairings) successfully merged out of 6095 (in 159 pairings) input.
## 3716 paired-reads (in 86 unique pairings) successfully merged out of 3894 (in 147 pairings) input.
## 6865 paired-reads (in 99 unique pairings) successfully merged out of 7191 (in 187 pairings) input.
## 4426 paired-reads (in 67 unique pairings) successfully merged out of 4603 (in 127 pairings) input.
## 4576 paired-reads (in 101 unique pairings) successfully merged out of 4739 (in 174 pairings) input.
## 6092 paired-reads (in 109 unique pairings) successfully merged out of 6315 (in 173 pairings) input.
## 4269 paired-reads (in 20 unique pairings) successfully merged out of 4281 (in 28 pairings) input.
head(mergers[[1]])
## sequence
## 1 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGCAGGCGGAAGATCAAGTCAGCGGTAAAATTGAGAGGCTCAACCTCTTCGAGCCGTTGAAACTGGTTTTCTTGAGTGAGCGAGAAGTATGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACTCCGATTGCGAAGGCAGCATACCGGCGCTCAACTGACGCTCATGCACGAAAGTGTGGGTATCGAACAGG
## 2 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGCCTGCCAAGTCAGCGGTAAAATTGCGGGGCTCAACCCCGTACAGCCGTTGAAACTGCCGGGCTCGAGTGGGCGAGAAGTATGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACCCCGATTGCGAAGGCAGCATACCGGCGCCCTACTGACGCTGAGGCACGAAAGTGCGGGGATCAAACAGG
## 3 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGGCTGTTAAGTCAGCGGTCAAATGTCGGGGCTCAACCCCGGCCTGCCGTTGAAACTGGCGGCCTCGAGTGGGCGAGAAGTATGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACTCCGATTGCGAAGGCAGCATACCGGCGCCCGACTGACGCTGAGGCACGAAAGCGTGGGTATCGAACAGG
## 4 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGGCTTTTAAGTCAGCGGTAAAAATTCGGGGCTCAACCCCGTCCGGCCGTTGAAACTGGGGGCCTTGAGTGGGCGAGAAGAAGGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACCCCGATTGCGAAGGCAGCCTTCCGGCGCCCTACTGACGCTGAGGCACGAAAGTGCGGGGATCGAACAGG
## 5 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGCAGGCGGACTCTCAAGTCAGCGGTCAAATCGCGGGGCTCAACCCCGTTCCGCCGTTGAAACTGGGAGCCTTGAGTGCGCGAGAAGTAGGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACTCCGATTGCGAAGGCAGCCTACCGGCGCGCAACTGACGCTCATGCACGAAAGCGTGGGTATCGAACAGG
## 6 TACGGAGGATGCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGTGCGTAGGCGGGATGCCAAGTCAGCGGTAAAAAAGCGGTGCTCAACGCCGTCGAGCCGTTGAAACTGGCGTTCTTGAGTGGGCGAGAAGTATGCGGAATGCGTGGTGTAGCGGTGAAATGCATAGATATCACGCAGAACTCCGATTGCGAAGGCAGCATACCGGCGCCCTACTGACGCTGAGGCACGAAAGCGTGGGTATCGAACAGG
## abundance forward reverse nmatch nmismatch nindel prefer accept
## 1 579 1 1 148 0 0 1 TRUE
## 2 470 2 2 148 0 0 2 TRUE
## 3 449 3 4 148 0 0 1 TRUE
## 4 430 4 3 148 0 0 2 TRUE
## 5 345 5 6 148 0 0 1 TRUE
## 6 282 6 5 148 0 0 2 TRUE
Ici, la première fusion de séquences est affichée, et permet de définir les séquences biologiques les plus abondantes.
Mismatch et Indel, respectivement le nombre de de bases différentes sur une même position et le nombre d’insertion ou délétion observées ; permettent de constater si la fusion est correcte ou non.
C’est le cas ici, où la fusion ne présente ni mismatch, ni insertion ou délétion.
Création d’un tableau de séquences
Permet de créer un tableau en se basant sur les échantillons et les séquences biologiques observées.
seqtab <- makeSequenceTable(mergers)
dim(seqtab)
## [1] 20 293
Le 20 représente le nombre de lignes, correspondant au nombre d’échantillons ; et le 293 représente le nombre de colonnes, correspondant au nombre d’ASV
Distribution de longueur des séquences
table(nchar(getSequences(seqtab)))
##
## 251 252 253 254 255
## 1 88 196 6 2
Permet de déterminer que une séquence contient 251 nucléotides, 88 en contiennent 253…
Suppression des chimères
seqtab.nochim <- removeBimeraDenovo(seqtab, method="consensus", multithread=FALSE, verbose=TRUE)
Les chimères sont le résultat de la fusion de fragments d’ADN ayant des origines différentes. De ce fait celles-ci sont amplifiées également et doivent donc être retirées.
“removeBimeraDenovo” est la fonction permettant la suppression de ces chimères ; tandis que “method=”consensus”” va permettre de conserver les séquences non-chimériques.
## Identified 61 bimeras out of 293 input sequences.
dim(seqtab.nochim)
## [1] 20 232
61 chimères ont été identifiées parmi les 293 séquences, et ont été supprimées du tableau, laissant seulement 232 colonnes dans celui-ci.
Calcul de la proportion de lectures conservées
sum(seqtab.nochim)/sum(seqtab)
## [1] 0.9640374
Environ 96% de lectures ont été conservées après filtrage.
Création d’un “tableau de suivi” du tutoriel
getN <- function(x) sum(getUniques(x))
track <- cbind(out, sapply(dadaFs, getN), sapply(dadaRs, getN), sapply(mergers, getN), rowSums(seqtab.nochim))
colnames(track) <- c("input", "filtered", "denoisedF", "denoisedR", "merged", "nonchim")
rownames(track) <- sample.names
head(track)
## input filtered denoisedF denoisedR merged nonchim
## F3D0 7793 7113 6976 6979 6540 6528
## F3D1 5869 5299 5227 5239 5028 5017
## F3D141 5958 5463 5331 5357 4986 4863
## F3D142 3183 2914 2799 2830 2595 2521
## F3D143 3178 2941 2822 2868 2553 2519
## F3D144 4827 4312 4151 4228 3646 3507
L’objectif de cette partie est de créer un tableau permettant d’observer les modification effectuées au sein des lectures le long de toutes les étapes précédentes. La légère diminution à chaque étape permet de prouver que les données se précisent au cours de celles-ci.
Attribution de taxonomie
Le but de cette étape est d’effectuer une comparaison avec des données existantes pour pouvoir définir une taxonomie par la suite.
taxa <- assignTaxonomy(seqtab.nochim, "~/dossier_1/silva_nr_v132_train_set.fa.gz", multithread=FALSE)
Ici, les données ont dû être téléchargées avant de pouvoir les utilisées, mais un lien peut normalement être utilisé également.
Vérification de la taxonomie
taxa.print <- taxa # Removing sequence rownames for display only
rownames(taxa.print) <- NULL
head(taxa.print)
## Kingdom Phylum Class Order Family
## [1,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales" "Muribaculaceae"
## [2,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales" "Muribaculaceae"
## [3,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales" "Muribaculaceae"
## [4,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales" "Muribaculaceae"
## [5,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales" "Bacteroidaceae"
## [6,] "Bacteria" "Bacteroidetes" "Bacteroidia" "Bacteroidales" "Muribaculaceae"
## Genus
## [1,] NA
## [2,] NA
## [3,] NA
## [4,] NA
## [5,] "Bacteroides"
## [6,] NA
Toutes les bactéries font partie de l’ordre des Bacteroidales, tandis que la famille la plus représentée est celle des Muribaculaceae.
Le genre n’a pas pu être identifié dans la plupart des cas mais cela est attendu lorsqu’une taxonomie est utilisée.
Contrôle de précision
Le but de cette étape va être de vérifier que le l’algorithme ait bien fonctionné via contrôle de l’échantillon blanc “Mock”
unqs.mock <- seqtab.nochim["Mock",]
unqs.mock <- sort(unqs.mock[unqs.mock>0], decreasing=TRUE) # Drop ASVs absent in the Mock
cat("DADA2 inferred", length(unqs.mock), "sample sequences present in the Mock community.\n")
## DADA2 inferred 20 sample sequences present in the Mock community.
DADA2 a identifié 20 séquences d’échantillons dans la communauté “Mock”, qui vont être examinées et vérifiées dans l’étape suivante.
Vérification des séquences d’échantillons “Mock”
mock.ref <- getSequences(file.path(path, "HMP_MOCK.v35.fasta"))
match.ref <- sum(sapply(names(unqs.mock), function(x) any(grepl(x, mock.ref))))
cat("Of those,", sum(match.ref), "were exact matches to the expected reference sequences.\n")
## Of those, 20 were exact matches to the expected reference sequences.
Cette étape permet de comparer les séquences retrouvées dans la communauté “Mock” avec celles de références, afin de s’assurer de l’exactitude de nos résultats.
Sur les 20 séquences identifiées par DADA2, 20 séquences correspondent aux séquences connues de la communauté “Mock”.
Cela permet de confirmer qu’il n’y a eu ni contamination ni erreur dans la totalité de nos échantillons.